Conheça nessa postagem a importância de que possui esse órgão tão importante e que a sua doação poderá ser o nascimento de uma nova vida para outra pessoa.
Imagine poder oferecer uma segunda chance de vida a alguém que enfrenta uma batalha desafiadora contra doenças graves do sangue, como é o caso das leucemias, por exemplo. Imagine a esperança brilhante que você poderia proporcionar a uma criança, um pai, uma mãe, ou um amigo nessa situação angustiante. A doação de medula óssea é uma jornada de amor, altruísmo e humanidade que pode transformar vidas de maneiras inimagináveis.
Este post explora a importância vital da doação de medula óssea, revelando os extraordinários benefícios que podem resultar de um gesto aparentemente simples. Vamos mergulhar fundo nesta jornada, desvendando os mistérios da medula óssea, destacando o processo de doação e compartilhando histórias inspiradoras de indivíduos que abraçaram o poder de doar vida.
Junte-se a nós nesta missão de conscientização, pois a cada doador se une a um elo crucial na corrente da esperança, e é o nosso compromisso compartilhar essa importante mensagem sobre a doação de medula óssea.
As principais doenças em que há necessidade de se realizar transplante e, consequentemente, pode precisar da doação de medula óssea são as leucemias agudas.
A leucemia é um grupo de doenças do sangue que se origina na medula óssea, onde as células sanguíneas são produzidas. Ela é caracterizada pelo desenvolvimento descontrolado de células brancas do sangue (leucócitos), que geralmente são responsáveis por combater infecções. No entanto, nas pessoas com leucemia, essas células não amadurecem normalmente e não realizam suas funções de maneira adequada.
Existem diferentes tipos de leucemia, mas duas categorias principais são a leucemia mieloide aguda (LMA) e a leucemia linfoblástica aguda (LLA). Ambas as formas da doença podem evoluir rapidamente e representar uma ameaça à vida. Para muitos pacientes com leucemia, o transplante de medula óssea torna-se uma opção crucial e, frequentemente, a melhor chance de cura. Aqui está o porquê:
1. Tratamento Intensivo para portadores de doença de Alto Risco O tratamento da leucemia geralmente envolve quimioterapia e, em alguns casos, radioterapia. Ao se realizar o diagnóstico também costuma ser avaliado outras características genéticas e moleculares que podem apontar para uma doença com risco elevado de não responder ao tratamento habitual, ou seja, pode não curar apenas com quimioterapia e daí necessitar de tratamento denominado de consolidação baseado no transplante alogênico, de um doador.
2. Recidiva da Doença: Mesmo após o tratamento inicial, a leucemia pode voltar, muitas vezes de forma mais agressiva. Para pacientes que tiveram recidiva da doença ou não responderam bem ao tratamento inicial, o transplante de medula óssea se torna uma opção crítica.
3. Substituição das Células Leucêmicas: O transplante de medula óssea envolve a substituição das células leucêmicas na medula óssea do paciente por células saudáveis de um doador compatível. Isso é essencial para permitir que o corpo produza células sanguíneas normais e eficazes.
4. A Importância da Doação: Encontrar um doador compatível é fundamental para o sucesso do transplante de medula óssea em pacientes com leucemia. Muitas vezes, os doadores compatíveis não são parentes próximos, o que enfatiza a importância do registro de doadores de medula óssea e da generosidade de indivíduos dispostos a doar.
5. Esperança de Cura: Para muitos pacientes com leucemia, o transplante de medula óssea oferece a esperança de uma cura duradoura. É um procedimento desafiador, mas a oportunidade de uma vida saudável e livre da doença é uma motivação poderosa para pacientes e doadores.
Portanto, a doação de medula óssea desempenha um papel vital na luta contra a leucemia, oferecendo uma chance real de recuperação e vida plena para aqueles que enfrentam essa doença devastadora. Cada doador potencial é uma luz brilhante de esperança para pacientes com leucemia e suas famílias, tornando a conscientização sobre a importância da doação de medula óssea uma missão de vida e amor.
Doenças Não Malignas que Necessitam de Transplante de Medula Óssea
Quando pensamos em transplantes de medula óssea, muitas vezes associamos isso ao tratamento de cânceres do sangue, como a leucemia. No entanto, a doação de medula óssea também desempenha um papel fundamental no tratamento de várias doenças não malignas, onde a medula óssea está comprometida ou não funciona adequadamente. Aqui estão algumas dessas condições:
1. Anemias Graves: Algumas formas graves de anemia, como a anemia aplástica, podem ser tratadas com transplante de medula óssea. Nesses casos, a medula óssea não produz células sanguíneas em quantidade suficiente, levando à anemia profunda e a problemas de coagulação. O transplante de medula óssea pode substituir a medula disfuncional por uma medula saudável que produza células sanguíneas normais.
2. Distúrbios Genéticos: Algumas doenças genéticas raras, como a talassemia e a doença falciforme, podem ser tratadas com transplante de medula óssea. Nessas condições, as células sanguíneas são afetadas por mutações genéticas que comprometem sua função. Um transplante de medula óssea pode fornecer células-tronco saudáveis que produzirão células sanguíneas normais.
3. Síndromes de Imunodeficiência: Algumas síndromes de imunodeficiência, como a síndrome de Wiskott-Aldrich e a síndrome de Job, afetam o sistema imunológico, tornando o paciente mais susceptível a infecções. O transplante de medula óssea pode substituir as células defeituosas do sistema imunológico por células saudáveis, restaurando a imunidade.
4. Síndromes de Armazenamento Lisossômico: Essas são doenças metabólicas hereditárias que afetam a função das organelas chamadas lisossomos nas células. Em alguns casos, um transplante de medula óssea pode ajudar a corrigir esses distúrbios, permitindo que as células funcionem normalmente.
Para pacientes que sofrem dessas condições não malignas e que não respondem ao tratamento convencional, ou ainda apresentam piora progressiva da contagem de células sanguíneas, um transplante de medula óssea pode representar uma chance de cura ou uma melhora significativa na qualidade de vida. Portanto, a doação de medula óssea ajudará também aqueles que enfrentam essas doenças não malignas, oferecendo esperança e uma oportunidade de recuperação.
A doação de medula óssea é um procedimento crucial que pode salvar vidas. Existem duas principais formas de realizar essa doação, e a escolha entre elas depende das necessidades do receptor e das circunstâncias. Vou explicar como funciona cada uma:
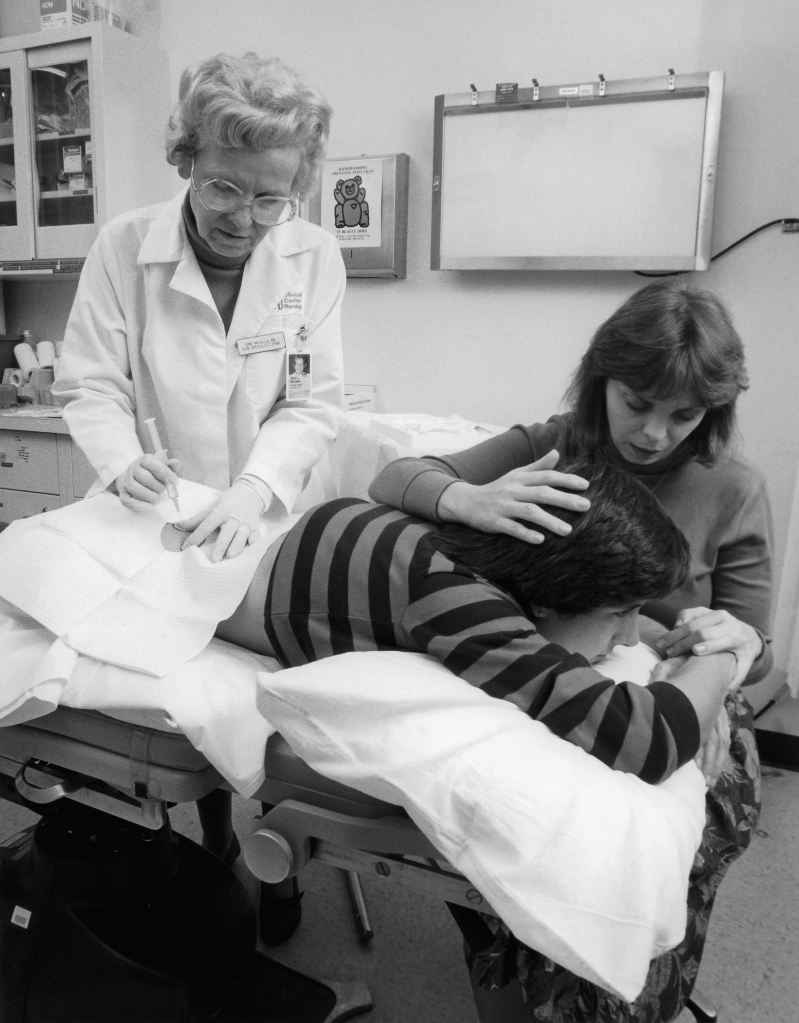
- Doação de Medula Óssea por Aspiração da Medula (Doador de Medula Óssea):
- Coleta da Medula Óssea: Nesse método, o doador é submetido a um procedimento cirúrgico chamado aspiração da medula óssea. Geralmente, a medula é retirada do interior do osso da bacia (osso ilíaco) por meio de agulhas. O doador é anestesiado durante o procedimento, o que torna a doação relativamente indolor. A cirurgia é realizada em ambiente hospitalar e leva cerca de 1 a 2 horas.
- Recuperação do Doador: A recuperação após a doação de medula óssea por aspiração pode levar alguns dias a algumas semanas. Os doadores geralmente sentem algum desconforto na área da incisão após a cirurgia, mas isso diminui com o tempo. A maioria dos doadores é capaz de retomar suas atividades normais dentro de algumas semanas.
- Cuidados Pós-Doação: Os doadores são acompanhados de perto pela equipe médica após a cirurgia para garantir uma recuperação adequada. É importante seguir as orientações médicas durante o período de recuperação.
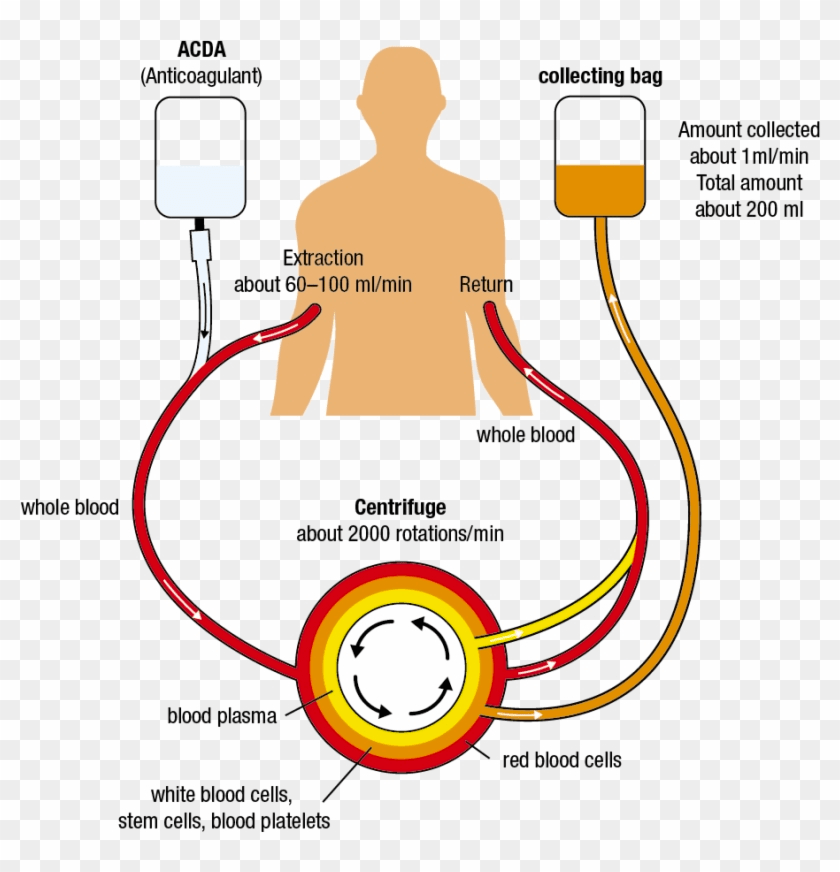
- Doação de Células-Tronco do Sangue Periférico (Doador de Células-Tronco do Sangue):
- Estimulação da Produção de Células-Tronco: Antes da doação, o doador recebe injeções de um medicamento que estimula a produção de células-tronco na medula óssea e as libera na corrente sanguínea.
- Coleta das Células-Tronco: Após a estimulação, o sangue do doador é coletado por um processo chamado aférese. Isso envolve a passagem do sangue através de uma máquina que separa as células-tronco do sangue periférico (plasma, glóbulos vermelhos, plaquetas, etc.). O sangue restante é devolvido ao doador.
- Recuperação do Doador: A doação de células-tronco do sangue periférico é um procedimento não cirúrgico e geralmente é bem tolerado. A maioria dos doadores se sente cansada durante o processo, mas a recuperação é relativamente rápida, geralmente em poucos dias.
- Cuidados Pós-Doação: Os doadores de células-tronco do sangue periférico também são acompanhados de perto pela equipe médica para garantir uma recuperação adequada. Eles podem ser aconselhados a evitar atividades físicas intensas por alguns dias.

A escolha entre a doação de medula óssea por aspiração e a doação de células-tronco do sangue periférico depende da recomendação médica, das necessidades do receptor e das circunstâncias específicas. Ambas as formas de doação são seguras e têm o objetivo de causar o mínimo de desconforto possível ao doador.
No Brasil, o processo de se tornar um doador de medula óssea é relativamente simples e pode ser feito seguindo estas etapas:
- Cadastro no Registro Nacional de Doadores de Medula Óssea (REDOME): O primeiro passo é se inscrever no Registro Nacional de Doadores de Medula Óssea (REDOME). O REDOME é uma base de dados que reúne informações de possíveis doadores de medula óssea no Brasil e é gerenciado pelo Instituto Nacional de Câncer (INCA).
- Critérios de Elegibilidade: Para se tornar um doador de medula óssea no Brasil, é necessário atender a alguns critérios, como ter entre 18 e 55 anos de idade, estar em bom estado de saúde e não ter algumas doenças graves. Além disso, é importante estar disposto a se comprometer com o processo, pois a doação de medula óssea é um compromisso de vida.
- Cadastro e Coleta de Amostra de Sangue ou Swab da Bochecha: Para fazer parte do REDOME, você pode se cadastrar de duas maneiras:a. Cadastro presencial: Você pode procurar um hemocentro ou banco de sangue em sua região para fazer o cadastro pessoalmente. Eles coletarão uma pequena amostra de sangue para fazer a tipagem HLA, que é usada para identificar doadores compatíveis.b. Cadastro online: Muitos hemocentros e organizações de doação de medula óssea também permitem o cadastro online. Nesse caso, você solicitará um kit de coleta em casa, que pode incluir um swab (cotonete) bucal. Após seguir as instruções e coletar sua amostra, você a enviará de volta para análise.
- Espera por um Chamado: Depois de cadastrado no REDOME, suas informações são inseridas no sistema, e você se torna parte do grupo de possíveis doadores. Se um paciente com uma doença que requer um transplante de medula óssea tiver características genéticas compatíveis com as suas, você poderá ser chamado para doar.
- Exames e Avaliação Médica: Se você for identificado como um possível doador compatível, você passará por exames médicos detalhados para garantir que está em boa saúde e apto para a doação.
- Doação: Dependendo das necessidades do paciente e da recomendação médica, você será informado sobre o tipo de doação que será realizada: aspiração de medula óssea ou coleta de células-tronco do sangue periférico, conforme explicado anteriormente.
- Apoio e Acompanhamento: Durante todo o processo, você será apoiado por uma equipe médica e receberá informações detalhadas sobre o procedimento de doação.
É importante ressaltar que a doação de medula óssea é um ato voluntário e altruístico. Ao se tornar um doador, você está oferecendo uma chance de vida a alguém que enfrenta uma doença grave. É uma maneira significativa de fazer a diferença na vida de outra pessoa.
Lembrando que as informações podem mudar com o tempo, então é sempre recomendável verificar as orientações e os requisitos atualizados junto aos órgãos de saúde e instituições de doação de medula óssea no Brasil, como o Instituto Nacional de Câncer (INCA) e os hemocentros regionais.

E o seu papel nisso tudo?
Uma Jornada de Esperança e Solidariedade
Tornar-se um doador de medula óssea é mais do que um ato de generosidade; é um gesto que pode proporcionar uma nova chance de vida a alguém que enfrenta uma batalha contra doenças graves do sangue e do sistema imunológico. No Brasil, o processo de doação é acessível e pode ser realizado por qualquer pessoa que atenda aos critérios de elegibilidade. Cada novo doador que se junta ao Registro Nacional de Doadores de Medula Óssea (REDOME) representa uma luz de esperança brilhante para aqueles que aguardam por um doador compatível.
As histórias de pacientes que tiveram suas vidas transformadas graças à generosidade de doadores são emocionantes e inspiradoras. A doação de medula óssea é uma jornada de solidariedade que une pessoas em uma missão nobre: oferecer uma nova oportunidade de vida a alguém em necessidade.
Portanto, considerar se tornar um doador de medula óssea é um ato de amor e altruísmo que pode fazer uma diferença profunda no mundo. Ao se cadastrar no REDOME, você se torna parte de uma rede de esperança, oferecendo uma dádiva valiosa que pode salvar vidas e restaurar a saúde de alguém que luta contra doenças desafiadoras.
Vamos juntos conscientizar mais pessoas sobre a importância da doação de medula óssea, compartilhando esta mensagem de solidariedade e esperança. Cada novo doador é um elo crucial na corrente da vida, e, juntos, podemos fazer a diferença, um ato de cada vez. Junte-se a essa missão e faça parte desta jornada de amor e compaixão. Afinal, a doação de medula óssea é, verdadeiramente, a doação da vida.